Science , Vol. 278, pp. 407-411 (October 17, 1997)
THE GENETICS OF AGING
by
Caleb E. Finch and Rudolph E. Tanzi
C. E. Finch, Neurogerontology Division,
Andrus Gerontology Center
and Department of Biological Sciences,
University of Southern California,
3715 McClintock Avenue
Los Angeles, CA 90089-0191
Voice: 213-740-1758
E-mail: cfinch@molbio.usc.edu
and
R. E. Tanzi, Genetics and Aging Unit
Massachusetts General Hospital
Neuroscience Center Building 149
Thirteenth Street
Charlestown, MA 02129
Voice: 617-726-6845
E-mail: tanzi@helix.mgh.harvard.edu
ABSTRACT
The role of genetics in determining lifespan is complex and paradoxical. While the heritability of lifespan is relatively minor, some genetic variants significantly modify senescence in mammals and invertebrates, with both positive and negative impact on the age-related disorders and lifespans. In certain examples, the gene variants alter metabolic pathways, which could thereby mediate interactions with nutrition and other environmental factors that influence lifespan. Given the relatively low degree and variable penetrance of genetic risk factors that appear to affect survival and health at advanced ages, lifestyle and other environmental influences may profoundly modify outcomes of aging.
Genes exert strong controls on the lifespan and patterns of aging. Yet, we know little of how humans live five times longer than cats; cats, five times longer than mice; mice, 25 times longer than fruit flies (1, 2), or why the onset of Alzheimers Disease (AD) often differs by many years in identical twins. Equally obscure is the role of genetics in the unprecedented increases of human life expectancy at advanced ages (1). To approach these puzzles, we must understand how the potential lifespan of an individual is determined by gene-environment interplay, which ultimately modulates the rates of molecular and cellular involution during aging. Clearly, individual humans are subject to genetic risks for age-related diseases, for example AD, cancer, diabetes, heart disease, and stroke. Other mammals share subsets of these age-related diseases, but we do not know causes of mortality in flies or worms (2). Since the incidence of diverse diseases accelerates exponentially with increasing age, it is difficult to critically resolve whether general age-related changes, such as the loss of skin elasticity and the slowing of reflexes, are mediated by the same mechanisms governing specific age-related diseases.
The Heritability of Lifespans is Small
A convergence of new and old data shows that the heritability of lifespans accounts for <35% of its variance in short-lived invertebrates, the nematode (3) and fruit fly (4), and in mammals, the mouse (5) and human (6, 7) (Table 1). Two studies of human twins attribute most (>65%) of the variance to non-shared (individually unique) environmental factors (6, 7). Twins reared apart share even less heritability of lifespan (7). We do not yet appreciate the nature or penetrance of environmental influences acquired during postnatal and prenatal phases, or even earlier in the life of the prezygotic oocyte (8).
Nonheritable variations in lifespan are also found in laboratory populations of inbred lines of nematodes, fruit flies, and mice. Within each inbred line, individuals show wide variations in lifespan which, expressed as the coefficient of variation, approximate those of outbred populations (Table 1). Moreover, inbred worms and flies, but also outbred human populations, display multiphasic changes in mortality rates during aging, such that mortality rates initially accelerate exponentially with advancing age (Gompertzian mortality), but then decelerate markedly among the last survivors (1, 2, 9). It remains possible that residual genetic variations contribute to this demographic (actuarial) heterogeneity in inbred laboratory animals (10).
Nonetheless, we do not depreciate the importance of genetics to the evolution of lifespans which is exemplified by the efficacy of artificial selection for longer or shorter lifespans in outbred fruit flies (2, 11, 12). Lifespan is considered by evolutionary biologists to be a statistical outcome of selections for the reproductive schedule (1, 11-16). Depending on the selection regime operating on a given population, the heritability of life spans, is like other quantitative life history parameters that show heritabilities ranging from negligible to nearly complete (11, 13). It is recognized from effects of diet restriction on lifespans of mice of different genotypes (2) that gene-environment interactions can greatly modify lifespans. Thus, transgenic approaches will be useful in identifying the dependency of genetic influences on lifespan in different host backgrounds. It is also conceivable that transgenic experiments could also evaluate if genes or alleles that influence lifespan within a given species may also modify the lifespans of other species. However, it seems unlikely that a few genes determine the 25-fold difference in lifespans between rodents and humans (2, 11, 15).
Gene Expression in Aging
Mutations dramatically modify the lifespan of fruit flies (11, 15, 17-19), and nematodes (15, 20-24) through mechanisms that may be shared by common effects on metabolism and gene expression. In fruit flies, enhancer-trap systems (25) detect transcriptional changes of exquisite specificity in their location and timing, for example the decreased expression of wingless (wg) is restricted to specific cells of the antennae (23). However, the relatively greater expression of heat shock hsp70 in flight muscles of old flies during stress responses is due to post-transcriptional age changes (17). In rodent brain and other tissues, selective changes in gene activity are also observed (2), such as the increased expression of the astrocyte cytoskeletal protein GFAP, which is due to increased transcription (26).
Experimental manipulations of life span in flies and rodents also cause corresponding changes in gene activity. During temperature-induced shifts in fruit fly life spans, the age-schedule for decreased expression of wg and engrailed (en) in the adult antenna (19) and for increased expression of hsp70 in muscle (17) is also shifted in proportion to lifespan over a three-fold range (Fig. 1). Accelerated age changes in gene expression occur in very short-lived mutants carrying drd, Hk1, or Sh5(19), and in catalase-null flies (17). These cell-specific age changes could not be detected in homogenates of whole flies, which are often used for biochemical studies of aging. Taken together, the anatomic and temporal specificity of changes during aging, and their scheduling in proportion to variations of lifespan in different situations, suggest the hypothesis that changes in gene activity during aging in flies are physiologically coordinated through humoral or neural factors. Although no data are available on how aging alters hormones or metabolites in the hemolymph of fruit flies, in certain castes of honeybees, there are increases of juvenile hormone that trigger senescence (2, 27). In view of the age changes in regulatory pathways involving en (transcription factor) and wg (secreted molecule mediating intercellular signaling), it will be of interest to examine expression of these and related genes in flies of different lifespans that were selected for early and late reproduction (11, 12).
The similar effects of very different mutations (drd, Hk1, or Sh5) on accelerating age changes in gene expression recalls the shared outcomes of defects in various genes that cause AD, all of which increase production of the amyloid b -peptide (Ab ) and lead to similar neurodegenerative changes at later ages. The potential influence of physiological factors in rat brain aging is shown by impact of food restriction, which slows the age-related increases in GFAP transcription and other markers of glial activation (26), while also increasing lifespan (2, 16, 18).
Gene expression is also altered during aging in bakers' yeast (Saccharomyces cerevisae), in which solitary mother cells sustain a finite number of asexual replicative cycles that, through budding, yield a limited number of progeny (28-31). Postreplicative yeast mother cells become enlarged, have changed optical properties (granularity), and may lyse during the next hours to days. Mutants selected for stress resistance (cold and nitrogen starvation) showed increased budding cycle lifespans on some genetic backgrounds, for example the SIR4-42 allele of the UTH2/SIR4 gene, which increased lifespan and stress resistance. The SIR complex (Silent Information Regulator) transcriptionally silences genes at telomeres and genes required for sexual mating (HML, HMR). The mutant SIR4-42 protein causes a redistribution of SIR proteins from telomeres to the nucleolus, as also occurs in senescent wild-type yeast cells (29, 30). Under- and over-expression of wild-type UT4 correspondingly increases or decreases the number of budding cycles. Mutation of the SGS1 gene, which encodes a DNA helicase with homology to the human gene for Werner's Progeria, also causes premature aging with redistribution of Sir3 to the nucleolus (30). The redistribution of telomeric silencing proteins is consistent with selective transcription of previously silenced genes, HMRa1 (29) and certain subtelomeric genes, such as URA3 (28). Other non-telomeric yeast genes also show altered expression during aging, for example decreased expression of the "longevity determining genes" LAG1 and RAS1 (28). The concentration of rRNA is lower in old yeast cells (28), possibly consequent to the redistribution of SIR proteins to the nucleolus. In certain mammalian brain neurons, the nucleolus shrinks at later ages through unknown mechanisms (32). In the filamentous fungus Podospora anserina, a long-lived mutant was mapped to a locus encoding grisea a copper-activated transcription factor implicated in the mtDNA instability that occurs during senescence in this species (31). Taken together, these findings suggest that selective changes of gene regulation have important roles in cell phenotypes of aging, as is widely found in development. However, it remains to be learned how many of the changes in gene activity during aging are secondary to other underlying causes, such as oxidative damage to molecules in the extracellular matrix as observed in mammals (2, 15, 18).
Budding cycle senescence in single yeast cells differs from the well-studied clonal (replicative) senescence of human diploid fibroblasts (33, 34) in which telomeres shorten during clonal senescence in vitro and in vivo (33). In yeast, telomeres do not shorten during budding cycle senescence (28), whereas induced telomere shortening appears to increase lifespan (29). A broad similarity, however, is selectivity of changes in transcription; so far, no examples of changes in homologous genes have come to light. In view of the increased resistance of mammalian fibroblasts to apoptosis during potentially prolonged postreplicative phases (33, 34), it will be of interest to learn details of cell involution and eventual lysis in postreplicative yeast mother cells.
Towards a Genetics of Longevity
Schächter, et al (35) proposed a three-part classification of candidate loci for longevity: (i) genes with homologues that influence longevity in other species; (ii) genes mediating cellular maintenance and repair; and (iii) genes associated with susceptibility to major age-related diseases.
(i) A search is underway to find genes associated with longevity in humans and other mammals, with a focus on the very elderly. In centenarians, the strongest candidate is the e2 allele of the apolipoprotein E (APOE) gene. The e4 allele, which promotes vascular disease, was 50% less frequent than in younger controls, while the frequency of e2, which is strongly associated with hyperlipidemia when homozygous, was higher in centenarians (36). Nonetheless, occasional centenarians with e4 /e4 are not demented (37). Italian centenarians show 50% lower frequency of apoB with low tandem repeats (apoB-VNTR) than in young controls, but this difference was not found in French or Finnish centenarians (38). The multigene major histocompatibility system (HLA in humans, MHC in mice) continues to spark interest as a source of longevity enhancing alleles. Mice carrying the H-2d allele have longer lifespans, increased immune vigor, and fewer lymphomas during aging (2, 5). Although centenarians show significant enrichment of up to two-fold in certain alleles of the HLA -A, -C, and -DR series (39, 40), no finding has yet been generalized across human populations.
Candidate longevity genes for humans are being found by searches for single gene mutations that extend the lifespan of the nematode Caenorhabditis elegans. Six induced mutations that extend life expectancy by 40 to 100% (20-23) share increased resistance to stressors including temperature, free radicals, and UV light. The first such mutation was age-1, which doubles maximum lifespan (20). Other nematode mutations also associate extended adult lifespan with stress resistance. The greatest increases in lifespan are associated with two genes that cause constitutive formation of the dauer larval stage (Table 1): age-l, identified with a phosphatidyl-inositol-3-OH kinase (22), and daf-2, with an insulin receptor-like gene (23). daf-2 activation by the dauer pheromone is hypothesized to be mediated by PIP-3 (23). This mechanism would be consistent with genetic evidence that daf-2 and age-1 participate in an epistatic pathway (21) and the greater lipid accumulation of daf-2 mutants (23). The regulation of nematode lifespan by insulin-like signaling is consistent with the extension of lifespan in rodents by food restriction (2, 18). Another set of long-lived mutants are those involving the clock gene (clk), which have slowed development, lengthened cell cycles, and modified adult behavior (24). clk-l encodes a short protein, an 82 amino acid tandem repeat which is highly conserved in eukaryotes; its yeast homolog CAT5/COQ7 indirectly regulates the transcription of genes that modulate energy metabolism to permit growth on nonfermentable carbon sources. The longest lived C. elegans are daf-2/clk-1 double mutants with a five-fold increase in lifespan (24). The increase of total life span by the dauer mutations daf-2 and daf-23 represents a two-fold increase in the adult phase in addition to the extended development.
(ii) A longevity locus for cellular maintenance is indicated by loss-of-function in the recently identified gene for the Werner's Syndrome, a rare autosomal recessive adult-onset progeroid with early manifestations of aging, such as hair loss, skin atrophy, premature heart disease, and various tumors. The Werner gene resembles the DNA (RecQ) helicases (41). Loss-of-function mutations in this gene lead to impaired DNA replication or DNA repair, resulting in the accumulation of various somatic DNA mutations and rapid decrease in telomere length (15). In contrast to the fruit fly mutations that accelerate multiple changes in gene expression, the Werner progeria mutations do not uniformly accelerate aging, e.g., there is no evidence for cognitive decline or AD. Nonetheless, because mutation of a yeast homologue also induced a progeroid phenotype (30), it is possible that that helicase functions could mediate a wider range of cellular aging changes than previously thought. A common polymorphism in the Werner progeria gene was recently associated with 2.7-fold higher risks of heart attacks (41), but the cellular mechanisms involved are not defined.
(iii) The third category includes genes involved in age-related neurodegenerative, cardiovascular, cancer, and immunological disorders. AD is an increasingly common neurological disorder of the elderly, which accounts for 70% of all cases of late-onset dementia (after 60 years of age) and currently causes greater than 100,000 deaths per year in the US (42). Because the incidence of AD doubles every five years after 60 years of age (42), the incidence of AD is expected to increase further as more survive to advanced ages. Characteristic neuropathological features of AD include neurofibrillary tangles (NFT) and extracellular deposits of Ab in senile plaques and cerebral blood vessels. NFT and Ab also accumulated to a lesser extent in individuals who reach advanced ages without clinical dementia (42). Although everyone might develop AD if they lived long enough, environmental risk factors most likely interact with genetic risk factors to determine age of onset. This argument can be extended to cancers, because carcinogenic determinants that vary with advancing age (greater than 30 years of age) appear to be distinct from those that are environmentally determined (43). The former may involve impaired DNA surveillance or activation of quiescent cells with damaged DNA. Meanwhile, impaired cell death (apoptosis), in combination with increased cellular proliferation, may underlie age-dependent decreases in the suppression of tumor formation due to an altered tissue microenvironment (44). The accumulation of replicatively senescent fibroblasts in vivo during aging could alter the cellular microenvironment by the increased secretion of proteases and other matrix-degrading enzymes (33).
The Genetics of Alzheimer's Disease and Cell Death
In contrast to the findings on lifespans, genetic effects are found in late-onset cognitive declines. In twins, the concordance with diagnosed AD (up to death) was two- to three-fold greater for MZ than DZ pairs (45). However, the onset of AD varied widely, with only 50% of concordant MZ or DZ pairs becoming demented within five years of the first (within-pair differences in onset ranged from 0 to 16 years). The estimates of AD that are attributed to known and undefined genetic risk factors show decreases at later ages. This trend parallels vascular disease risk factors in twins and diminishing genetic influences on blood lipids at later ages (46).
Although most AD occurs in populations older than 60 years, approximately 5% arises before 60 years of age (47), when it is frequently clustered in families [early onset Familial AD (FAD)]; this is autosomal dominant and virtually 100% penetrant (42). These genetically heterogeneous conditions involve defects in at least three different genes and lead to indistinguishable neuropathology. The major component of brain amyloid in AD brains is a 4-kD peptide (Ab ) derived during the processing of the amyloid b -protein precursor (APP) (42). Many missense FAD mutations in the APP gene cluster around the Ab domain and increase the production of Ab . Transgenic mice with APP mutations accumulate Ab in the brain, but without neuronal loss or NFT (42, 48). This result suggests that Ab deposits may be necessary, but are not sufficient to yield AD-like neuropathology, at least in the mouse brain. Ongoing studies may show whether other AD components such as human forms of the tau protein, a main component of NFT, or of human forms of genes expressed in glia are required to induce AD in mice. In transgenic mice over expressing wild-type APP, early death and brain abnormalities occurred in the absence of Ab deposits. Since early death also occurs in 20% of nontransgenic mice of this strain (FVB/N), APP overexpression may accelerate a natural, age-related brain disorder (49).
Mutations in APP and in the presenilins, PS-1 and PS-2, account for 50% of the occurrence of FAD (42). The vast majority of FAD mutations occur in PS-1, each being atypically restricted to single family (42). Despite this genetic diversity, AD may nonetheless deve1op from a broadly shared pathogenic process. Most FAD mutations in APP, PS-1, and PS-2 increase the secretion of Ab , by favoring the production of 'long' Ab , a form of the peptide with 42 rather than 40 residues, which is more prone to aggregations and to forming Ab deposits in the brain (42). These data argue for a central role of Ab 42 in amyloid deposits of AD brains. Moreover, the APOE-e4 allele is associated with increased Ab load in AD brains (42, 50). The e4 allele is associated with late-onset AD and stroke due to Ab deposits in cerebral blood vessels (51). The risk for AD conferred by e4 is greatest for onset between 61 and 70 years of age (47). Meanwhile, the e2 allele is associated with a decreased risk for AD, which is consistent with its association with longevity (36).
Although functions of the presenilins are unknown, they have 50% similarity to the C. elegans protein SEL-12 which is a facilitator of the LIN-12 Notch receptor. Moreover, the death of PS1-null mouse embryos from defects in somite segmentation indicates that presenilins have key roles of the presenilins in axial skeleton development (52). Presenilins are also substrates for a caspase-3 family protease during induced apoptosis. Moreover, the PS2-N141I FAD mutation confers increased susceptibility to apoptosis and increased cleavage of the PS-2 protein by caspase-3 (53). Caspase 3-generated presenilin fragments could, in turn, also make cells more vulnerable to apoptosis. Apoptosis in aging can either be beneficial by eliminating dysfunctional cells so that they can be replaced by proliferation (homeostasis), or detrimental by eliminating irreplaceable cells such as neurons, leading to neurodegeneration. For example, in aging rodents, D2-dopamine receptor-containing neurons are lost through apoptosis, whereas apoptosis decreases the accumulation of nonfunctional T-cells during food restriction (54).
Apoptotic death of neurons during AD (55, 56) may result from impaired energy metabolism and the enhanced generation of 4-hydroxynonenal (HNE)(56), a key mediator of neuronal apoptosis induced by oxidative stress. Alternatively, neuronal death in AD could be due to enhanced caspase-mediated cleavage of the presenilins in association with increased vulnerability to apoptosis (53) or to altered interactions of presenilins with concatenin proteins (57). The concatenins also interact with edematous polyposis coli (APC) tumor suppressor, the inactivation of which can lead to increased proliferation or inhibition of apoptosis owing to transcriptional activation mediated by b -catenin (58). Mutations in either APC or b -catenin occur in one-third of melanomas (59). Thus, b -catenin may become oncogenic when mutated or upregulated by inactivation of APC. Since b -catenin can also interact with the presenilins, endoproteolysis of PS-1 or PS-2 may serve to regulate proliferative vs. apoptotic signals mediated by catenins. The involvement of the catenins in the Wingless signaling pathway of fruit flies, which is mutually inhibitory with Notch (60), may explain how a nematode presenilin homolog, SEL-12, facilitates the LIN-12 Notch receptor.
Alternate Life Histories
Genetic variants in the invertebrate models described above show that altered scheduling of aging can arise from induced mutations, with great shortening or lengthening of lifespans. Moreover, environmental factors may determine alternate life histories within the same population. An instructive example is the l0-fold difference in lifespans of female worker bees (Apis mellifica), which have rapid senescence and lifespans of months, whereas queens of the same genotype show much slower senescence during lifespans of many years of active egg production. These alternate life histories in females are determined by exposure of larvae to nutrients and juvenile hormone.
Brown trout (Salmo trutta) also have several coexisting life histories, with the giant ferox trout living more than five times longer than smaller sized trout (2, 61). At a critical body size, some adult brown trout switch from plankton feeding to piscivory, which allows faster growth and, contrary to predictions from food-restriction theory (2, 18), a much longer lifespan (61). A possible genetic basis is the much higher prevalence of a Lactic DeHydrogenase allele (LDH-5) in ferox than in the small, short-lived brown trout in the same local population. In the killifish (Fundulus heroclitus), geographic subpopulations differ in LDH alleles and isozyme activities that were shown experimentally to alter rates of development (62).
When these examples are considered together with the C. elegans mutations that modify metabolism and lifespan, it seems plausible that metabolic regulation may be a general feature of life history variants. These examples are also consistent with the evolutionary theory of life histories, in which selection for the duration and frequency of reproduction indirectly modifies the potential lifespan (11, 13-15). The extensive plasticity in adult lifespans is also represented in the recent major increases of human lifespan, although it is not obvious how this increase could have resulted from natural selection.
Conclusions
During recent centuries, technological advances that allow a higher quality of life and general health have revealed an untapped potential of the human genome to support major increases in life expectancy at all ages (1). Nonetheless, the explosive increase of cancer, vascular disease, and AD at later ages were not widely experienced in historical human populations, so that there was little selection against these diseases at their present age of occurrence (2, 11, 14, 15). Ironically, genetic tools that were developed in the various genome projects now allow easy identification of genetic risk factors such as those for conditions that may comprise health in later years. One might consider that, if the human life expectancy should approach the present record of 122 years, some existing gene polymorphisms which may be "lying in wait" could rise up to challenge the quest for health at advanced years. The growing inventory of genetic and environmental factors affecting the lifespan, along with an understanding of gene-to-gene and gene-environment interactions, should arm us well for the continuing battle with morbidity. Genetic studies of age-related human diseases and longevity mutants of animal models have identified many risk factors that affect metabolism and resistance to stress. Future studies of animal and cell models of aging should identify many paths, some of them convergent, by which environmental factors modify genetic risk factors in aging. The relatively minor heritability of human lifespan at advanced ages and the variable penetrance of genetic risk factors implies that choice of life style profoundly influences the outcomes of aging (63).
Table 1:
Heritability and Variance Characteristics of Lifespans in Selected Vertebrates and Invertebrates.|
Species |
Heritability of Lifespan* |
Coefficient of Variation of Lifespan § |
Lifespan (Mean) |
|
Nematode (3) |
15 days (25°C) |
||
|
within line |
0 (self-fertilizing) |
34% |
|
|
between lines |
0.34 |
19% (16-24%) |
|
|
Flies |
|||
|
Fruit fly, inbred lines (4) |
40 days (25°C) |
||
|
within line |
[<0.01] |
||
|
between lines |
0.06-0.09 |
11% |
|
|
Medfly, outbred (9) |
not determined |
45% |
21 days (25°C) |
|
Mouse, inbred lines (5) |
27 months |
||
|
within line |
[<0.01] |
24% (18-71%) |
|
|
between lines |
0.29 |
16% |
|
|
Human twins (6, 7) |
72 years |
||
|
0.23-0.33 |
MZ, 19%; DZ, 25% |
* A linear model partitioned the heritability of lifespans into additive, dominant, and epistatic components (3-7). For animal models, values are "narrow sense heritability" that represents the additive component (64). For human twin monozygous (MZ) and dizygous (DZ) pairs from Denmark (6) and Sweden (7), ennvironmental terms were included in the model (64); the values show the range of heritability in both studies. Danish twins (6) showed the best fit with a model of dominant heritability and non-shared environment.
§ The coefficient of variation for lifespan is the standard deviation of lifespan in the population as a percent of the mean lifespan (65). Variations of lifespan within inbred lines could represent the microenvironment (66), but also residual genetic variations (10, 64).
References and Notes
1 . J. Vaupel, in Between Zeus and the Salmon: The Biodemography of Aging, K. Wachter, C. E. Finch, Eds. (National Academy of Sciences, Washington, D. C.; 1997).
2. C. E. Finch, Longevity, Senescence, and the Genome (University of Chicago Press, Chicago, Illinois; 1990).
3. T. E. Johnson and W. B. Wood, Proc. Natl. Acad Sci. U.S.A., 79, 6603 (1982).
4. D. E. L. Promislow, M. Tater, A. A. Khazaeli, and J. W. Curtsinger, Genetics, 143, 839 (1996).
5. R. Gelman, A. Watson, R. Bronson, and E. Yunis, ibid, 118, 693 (1988).
6. A. M. Herskind, et al., Hum. Genet., 97, 319 (1996).
7. E. Ljungquist, S. Berg, J. Lanke, G. E. McClearn, and N. L. Pedersen, in Aging and Survival: Studies of Social, Biobehavioral, and Genetic Correlations (Dept. Geriatric Medicine, University of Goteborg, Sweden).
8. In humans, the unfertilized egg typically exists for twenty or more years as a cell in the mother's ovary, which because of its origin before her birth, could carry influences from the matrilineal grandmother (C. E. Finch and J. C. Loehlin, Behav. Genetics, in press). Prefertilization influences in mammals are less likely for sperm, which are generally short-lived. However, social insect queens store sperm for many years.
9. J. R. Carey, et al., Science, 258, 457 (1992).
10. A caveat is that variations in lifespan observed in certain inbreeding situations could represent residual genetic variance. Brother-sister inbreeding can achieve <0.001 residual genetic variance, but may never reach "0" because of point mutations and chromosomal rearrangements, including unstable mobile genetic elements (P-elements, flies; retroviruses, mice) and expansion-contraction of trinucleotide repeats. Self-fertilizing nematodes more readily approach isogenicity (3), although spontaneous mutations could still lead to genetically distinct subpopulations that would be hard to detect. Mitochondrial DNA replication is more error-prone and could also contribute variations to chromosomally isogenic strains.
11. M. R. Rose, The Evolutionary Biology of Aging (Oxford U. Press, New York; 1991).
12. M. R. Rose, in Ref. 1.
13. D. A. Roff, The Evolution of Life Histories: Theory and Analysis (Chapman & Hall, New York; 1992); S.C. Searns, The Evolution of Life Histories (Oxford University Press, New York; 1992).
14. S. Tuljapurkar, in Ref. 1.
15. G.A Martin, S. A. Austad, and T. E. Johnson, Nature Genet., 13, 25 (1996).
16. J. Carey, in Ref. 1.
17. J. Tower, BioEssays, 18, 799 (1996); J. C. Wheeler, E. T. Bieschke, J. Tower, Proc. Natl. Acad. Sci. U.S.A. 92, 10408 (1995).
18. R. S. Sohal and R Weindruch, Science, 273, 59 (1996).
19. B. Rogina and S. L. Helfand, Mech. Of Aging and Devel., 63, 89 (1997); B. Rogina, S. Benzer, S. L. Helfand, Proc. Natl. Acad Sci. U.S.A., 94, 6303 (1997).
20. D. B. Friedman and T. E. Johnson, Genetics, 118, 75 (1988).
21. P. L. Larsen, P. S. Albert, and D. L. Riddle, Genetics, 139, 1567 (1995); C. Kenyon et al., Nature, 366, 461 (1993).
22. J. Z. Moms, H. A Tissenbaum, and G. A. Ruvkun, Nature, 382, 536 (1996).
23. K.K Kimura, H.A Tissenbaum, Y. Liu, and G. Ruvkun, Science, 277, 942 (1997).
24. B. Lakowski and S. Hekimi, Science, 272, 1010 (1996); J. J. Ewbank, et al., Science, 275, 980 (1997).
25. Enhancer-trap systems are random inserts used to identify genes that change in transcriptional activity. Flies are engineered to contain a single insertion of a P-type transposable element with a weak promoter and a reporter gene whose product generates a histochemically detected product. When the nearby gene becomes active, the trapped reporter may be detected and localized to particular cells. Insertional genetic techniques, such as required for enhancer-trapping and to increase gene copy number for testing hypotheses about aging (16, 22), may induce position effects that may haphazardly modify lifespan and details of aging [M. Kaiser, M. Gasser, R. Ackerman, and S. C. Stearns, Heredity, 78, 1 (1997)].
26. T. E. Morgan, et al., Free Rad. Biol. Metab., 23, 524 (1997).
27. T. Giray, G. E. Robinson, Proc. Natl. Acad. Sci. USA, 15, 11718 (1996); S. E. Fahrbach and G. E. Robinson, Dev. Neurosci., 18, 102 (1996).
28. S. M. Jazwinski, Mol. Microbiol., 4, 337 (1990); S. M. Jazwinski, Science, 273, 54 (1996); S. P. Kale and S. M. Jazwinski, Dev. Genet., 18, 154 (1996); S. Kim, B. Villeponteau, S. M. Jazwinski Biochem. Biophys. Res. Commun., 219, 370 (1996).
29. T. Smeal, et al., Cell, 84, 633 (1996).
30. B. K. Kennedy, et al., Cell, 89, 381 (1997); D. A. Sinclair, K Mills, and L. Guarente, Science, 277, 1313 (1997).
31. D. Osiewacz and U. Nuber, Mol. Gen Genet., 252, 115 (1996).
32. C. E. Finch, Trends Neurosci., 16, 104 (1993).
33. J. Campisi, Eur. J. Canc., 33, 703 (1997); J. R Smith, O. M. Periera-Smith, Science, 273, 70 (1996).
34. E. Wang, Can. Res., 55, 2284 (1995).
35. F. Schächter, D. Cohen, T. B. L.. Kirkwood, Hum. Genet., 91, 519 (1993).
36. F. Schächter, et al., Nature Genet., 6, 29 (1994).
37. E. Sobel, et al., Neurology, 45, 903 (1995).
38. G. DeBenedictis, et al., Hum. Genet., 99, 312 (1997).
39. J. Proust, et al., Tissue Ant., 19, 168 (1982); J. S. Thompson, et al., J. Am. Geriatr. Soc., 32, 274 (1984); H. Taketa, et al., Lancet, II, 824 (1987).
40. Yong-Xing, et al., Mech. of Aging and Dev., 94, 191 (1997).
41. C. E. Yu, et al., Science, 272, 193 (1996); C. E. Yu, et al., Amer. J. Med. Genet., 68, 494 (1997).
42. Alzheimer Disease, R. D. Terry, R. Katzman, K. L. Bick Eds. (Raven Press, New York; 1994); R. E. Tanzi, et al., Neurobiol. Dis., 3, 159 (1996).
43. D. Benson, N. Mitchell, and D. Dix, Mutat. Res., 356, 209 (1996).
44. K. D. McCullough, W. B. Coleman, G. J. Smith, and J.W. Grisham, Cancer Res., 57, 1807 (1997).
45. M. Gatz and N. L. Pedersen, Alzheimer Res. 2, 229 (1997); M. Gatz, et al., Journal Gerontol., 52A, M117 (1997).
46. D. A. Heller, N. L. Pedersen, U. DeFaire, G. E. McClearn, Am. J. Hum. Genet., 55, 1255 (1994).
47. D. Blacker, et al., Neurology, 48, 139 (1997).
48. D. Games, et al., Nature, 373, 523 (1995); K. Hsiao, et al., Science, 274, 99 (1996).
49. K. Hsiao, et al., Neuron., 15, 1203 (1995).
50. B. T. Hyman, et al., Proc. Natl. Acad. Sci. USA, 92, 3586 (l995).
51. E. H. Corder, et al., Science, 261, 921 (1993); S. M. Greenberg, et al, Ann. Neurol., 38, 254 (1995).
52. P. Wong, et al., Nature, 387, 288 (1997); J. Shen, et al., Cell, 89, 629 (1997).
53. T. Kim, W. H. Pettingell, Y.-K. Jung, D. M. Kovacs, and R. E. Tanzi, Science, 277, 373 (1997).
54. T. Shinkai, L. Zhang, S. A. Mathias, and G. S. Roth, J. Neurosci. Res., 47, 393 (1997); C. C. Spaulding, R. L. Walford, and R. B. Effros, Mech. of Aging and Dev., 93, 25 (1997).
55. J. H. Su, et al., Neuro Report, 5, 2529 (1994); G. Smale, et al., Exp. Neurol., 133, 225 (1995).
56. I. Kruman, A. J. Bruce-Keller, D. Bredesen, G. Waeg, and M. P. Mattson., J. Neurosci., 17, 5089 (1997).
57. Zhou, et al, Neuro Report, 8, 1489 (1997).
58. V. Korinek, et al, Science, 275, 1784 (1997); P. J. Morin, et al., ibid, 1787 (1997).
59. B. Rubinfield, et al, Science, 275, 1790 (1997).
60. J. D. Axelrod, et al, Science, 271, 1826 (1996).
61. A. Ferguson, J. B. Taggart, Biol. Journal Linn. Soc. 43, 221 (1991); M. Mangel., Evol. Ecol., 10, 249 (1996).
62. P. M. Schulte, M. Gomez-Chiarri, D. A. Powers, Genetics, 145, 759 (1997); L. DiMichele, M. E. Westerman, Mar. Biol., 128, 1 (1997).
63. These projects were supported by grants to C.E.F. from the N.I.A. and to R.E.T. from the N.I.A., N.I.N.D.S., and the Metropolitan Life Foundation. T. Johnson, G. McClearn, R. Miller, M. Tater, and J. Tower gave helpful comments.
64. The value for nematodes is the average of calculations from three different experimental paradigms, each replicated (3).
65. The coefficient of variation is a dimensionless number, calculated as [S.D./X] x 100 for each species, where S.D. is the Standard Deviation for lifespans in that population and X is the mean lifespan: nematode Caenorhabditis elegans, Bristol hermaphrodites (mean, 3 experiments) and mean of Bristol, Bergerac, and F2 (6 lines) (calculated by T. E. Johnson, pers. comm., from Ref. 3.); fruit fly (Drosophila melanogaster), 25 genotypes, each identically heterozygous for chromosome 2 (calculated by M. Tater, pers. comm. from Ref. 4.; medfly (Ceratitis capitata), (calculated by J. Carey, pers. comm., from Ref. 9.); mouse (Mus musculus), 20 inbred lines (calculated by R. A. Miller, pers. comm., from Ref. 5.); human twins, Danish born 1870-1880, with genders averaged to compute grand means for MZ and DZ twin pairs (63).
66. Variations in lifespans must also depend on local conditions. For example, total lifespans of C. elegans can be more than three-fold longer if a lack of nutrients during development triggers the dauer larval stage; the dauer may last 70 days without altering the adult phase, once food becomes sufficient (2, 20-23). Social and reproductive interactions can also increase mortality risk in flies and mice. The present sampling of populations under protected conditions nonetheless have similar variations in lifespan that give a sense of baseline variations. Also, see Notes 10 and 25.
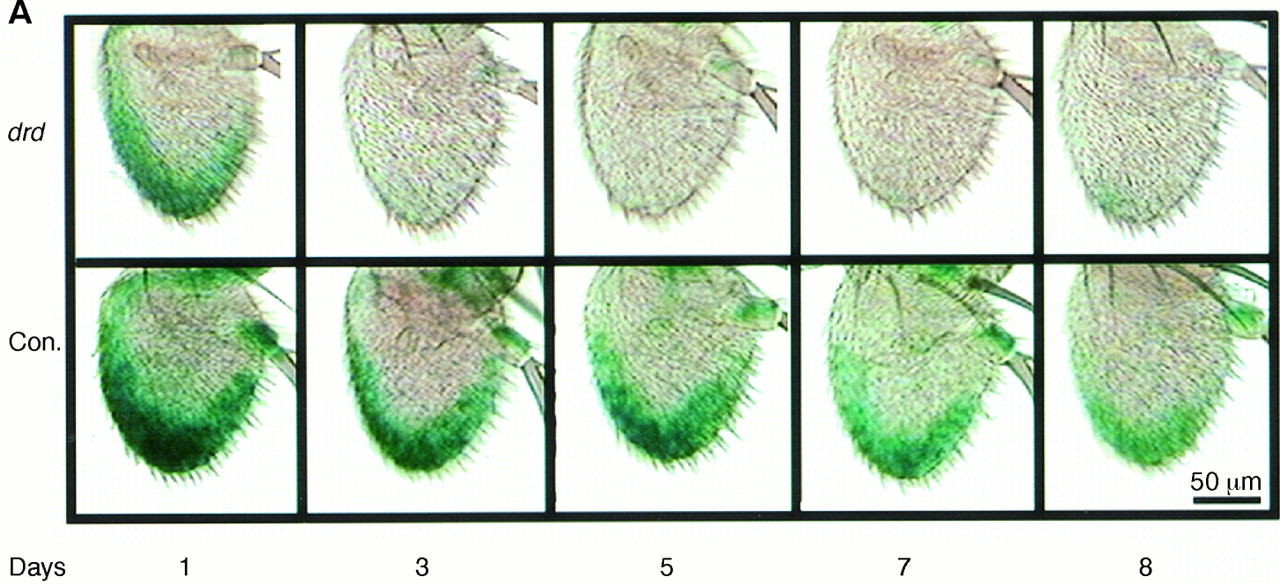
Figure 1:
Expression of the wg in Fruit Flies During Aging Changes in Proportion to the Lifespan.
A. The green color* shows the reporter signal from an enhancer-trap line in control flies (lifespan, 40 days) and in the short-lived mutant drd (lifespan, 6 days).
B. Expression of wg decreases gradually during aging in control antennae and more rapidly in drd.
C. Expression of wg as a percent of lifespan. Photographs provided by S. Helfand (from Ref. 19).
_______________________________
*[Editor's Note: Color information that was previously lost in the black-and-white copier, has now been scanned-in and posted in color.]