Stanley R. Primmer
1. Those individuals who are now approaching the maximum known human lifespan should be thoroughly studied both before and after they die (e.g., the oldest known woman, Jeanne Calment, now 122-years-old and who just celebrated a quiet birthday in her French nursing home last Friday, and the oldest known man, Thomas Mortensen, a 114-year-old American from San Rafael, California). The exact cause of death should be determined with precision. Data should be obtained for telomere length, percentage of cells which are senescent, percentage of 8-hydroxy-guanine content of mitochondrial DNA, percentage of mitochondrial DNA deletion, oxidative stress, antioxidant levels, rate of apoptosis, and percentage of cells undergoing mitotic division. While the individuals are living they should be interviewed concerning their life style, diet, use of nutritional supplements, use of tobacco, age of siblings and parents at time of death, and many other relevant factors. I propose that a scientific demographic research consortium should be organized to obtain and process these data. Geron Corporation could be enlisted to perform the telomere analysis and determine the percentage of senescent cells. Takayuki Ozawa and his colleagues at the University of Nagoya would be ideal to analyze mitochondrial damage. Prof. Bruce Ames of the University of California at Berkeley could also be involved in the mitochondrial analysis. Pantox Laboratories and/or Genox Corporation could analyze oxidative stress and antioxidant profiles. Immunosciences Lab could determine the rate of apoptosis and the percent of cells undergoing mitotic division. Other individuals or organizations could be involved instead of or in addition to those named. Additional analyses might also be desired.
[NOTE: This topic is almost identical to the first item on Dr. L. Stephen Coles's Top Ten List. It contains some
supplementary ideas, and is listed first to emphasize the urgency in obtaining data while the subjects are still alive. It
is suggested that the proposed scientific demographic consortium should be organized immediately.]
2. What is the mechanism by which telomere shortening causes replicative senescence?
The known sequence of genes in the cell senescent pathway includes ATM, p53, Ceramide, CAPP and/or p21 (=
WAF1/CIP1), and Rb (Elledge, pp. 1666, 1669; Savitsky, et al, p. 1752; Shay, pp. 381-382; Wright and Shay, p.
194; Shay, et al, 1991, pp. 5-6; Hannun, pp. 1855-1856; El-Deiry, et al, pp. 1169-1174; Canman, et al, pp.
280-283; Sherr, p. 1675; Enoch and Norbury, pp. 426-430; Lavin, et al, pp. 382-383). Ceramide and CAPP lie
between p53 and Rb in the processing pathway; likewise p21 also lies between p53 and Rb, but the relationship
between p21 and Ceramide or CAPP is unclear. The complete genetic pathway from an unknown gene or genes
upstream from the ATM and p53 genes needs to be defined. How does telomere shortening activate this unknown
gene(s)? Two possibilities are (1) "shortened telomeres are interpreted by the cells as DNA damage, signaling
induction of p53 and cell cycle arrest," and (2) the invoking of "changes in expression of regulatory genes located in
subtelomeric heterochromatin upon aging" (Shay, p. 382). Senescent cells are metabolically active, have some genes
which respond to mitogens and some which don't, resemble terminally differentiated cells, and are resistant to
apoptosis (Campisi, 1996, p. 498). How and why are senescent cells different from terminally-differentiated-or
postmitotic-cells and quiescent cells?
3. Does the accumulation of senescent cells in an organism contribute to the aging of that organism?
We do not age or die because our cells can no longer divide (Hayflick, p. 132; Fossel, pp. 52-53). Even cells from very old humans can still divide. Although senescent cells are still metabolically active (Campisi, 1996, p. 498), their biochemical functions have altered, and they can no longer perform as younger cells do (Hayflick, p. 132; Campisi, 1996, p. 499). This altered function "may have a substantial impact in aged tissue. . . . Moreover, relatively few senescent cells would be needed for some of these effects" (Campisi, 1996, p. 499).
The accumulation of certain types of senescent cells can have fatal results. Some centenarians have been shown to
have senescent CD8 cells (Effros, et al, p. F20), which could be part of the basis for immunological senescence,
leaving the elderly vulnerable to disease and infection. Loss of replicative capacity and alteration in gene expression
of senescent arterial cells have been implicated in age-related cardiovascular disease (Chang and Harley, pp.
11190-11193). These are two examples in which the effects of aging are caused by the fact that senescent cells can
no longer replicate. What other symptoms of aging are the result of accumulation of senescent cells?
4. Does shortening of telomeres cause changed expression of some genes, or is this caused by other concurrent factors? What is the mechanism by which change of gene expression occurs in senescent cells?
A number of researchers have observed the change in gene expression of senescent cells as compared to younger
ones (Hayflick, pp. 132-133, 135; Shay, et al, 1992, pp. 486-489; Campisi, 1996, p. 499; Dimri, et al, p. 9363;
Linskens, et al, pp. 3244, 3249-3251; Kipling, p. 137; Fossel, pp. 73-82). Some of the theories to explain this effect include:
d. Possibly some changes in gene expression of senescent cells are due to mutations caused by free radicals. Nuclear DNA would seem to be more vulnerable to free radical attack during mitosis when it is not protected by the nuclear envelope. (However, bundling of the chromatin fiber into the karyotypic chromosome form at this time might afford some protection.) If this hypothesis is valid, one might expect some correlation between the number of cell divisions and the number of mutations caused by free radicals. This view is entirely speculative on my part. Work by Linskens, et al, showed senescent changes in cells which began in vitro as neonatal cells (Linskens, et al, pp. 3244-3251). One would think that these cells would have had more limited exposure to free radicals in vitro than comparable cells in vivo over a longer time period. The consistent repetition of specific phenotypic characteristics (Shay, et al, 1992, p. 489) also indicates that at least some senescent changes in gene expression are probably not due to mutations, which could be expected to be more random in nature.
Change in gene expression may be due to multiple factors, including shortening telomeres and free radical induced mutations. Those changes which are affected by telomere length may be caused by a combination of heterochromatin, telomere binding proteins, and modification of the physical configuration of telomeric chromatin, as described above. Kipling describes telomere function and structure as follows:
Protecting a telomere from fusion and recombination may not be a direct result of the DNA sequence but rather a result of the action of telomere binding proteins. In S. cerevisiae the RAP1 protein, in concert with a range of other factors, is speculated to form a heterochromatin-like domain at the telomere, making the double-stranded terminus inaccessible to nucleases or recombination enzymes as well as ensuring that natural chromosome ends do not activate cell cycle checkpoints which monitor DNA damage (Kipling, p. 97).
Alteration of gene expression begins to occur prior to cellular senescence. Hayflick expressed the opinion that the
same process which causes aging affects both dividing and nondividing cells (Hayflick, pp. 133-134). If he is
correct, then cellular aging as it is related to shortening telomeres is a secondary effect. However, b-galactosidase
has been found to occur in replicatively senescent cells, but not in quiescent, terminally differentiated, or immortal
cells (Dimri, et al, pp. 9364-9367; Campisi, 1996, p. 499). This is only one example of an effect in senescent cells
which is different from other cells. Other phenotypical changes in senescent cells include: overexpression of
collagenase, interleukin-1, and cytokines; underexpression of collagenase inhibitors and interleukin-6 (Campisi,
1996, p. 499); repression of c-fos (Campisi, 1992, pp. 398-400); and modified expression of terminin (Wang, p.
422). Cristofalo et al compare differential gene expression in young and senescent cells (Cristofalo, et al, pp.
429-432). Linskens et al list several genes which are differentially expressed in senescent cells (Linskens, et al, p. 3248).
The complete molecular structure of human telomeres with binding proteins needs to be characterized as it has been
in Oxytricha nova (Schultz; see also Kipling, pp. 97-107). The physical conformation of both single-strand and
double-strand telomere binding proteins with the telomere should be defined in detail. A mammalian double-strand
telomere binding protein, TRF1, has been identified (van Steensel and de Lange, pp. 740-743; Zhong, et al, pp.
4834-4843), but not a single-strand binding protein.
5. How can telomeres be extended in all somatic cells of laboratory animals and maintained at a constant length?
Would maintaining telomere length in somatic cells of laboratory animals extend their maximum life span?
Harley et al stated that ". . . if manipulation of cellular life span were technically possible through regulation of
telomerase, it might be possible to decrease morbidity or increase life span of the organism" (Harley, et al, p. 381).
Would laboratory animals in which telomere length is maintained have a higher incidence of cancer? The possibility
of uncontrolled proliferation of cells which never reach replicative senescence due to maintaining telomeres at a
more or less constant length should be considered. Could oncogenes promote cell replication which would result in
cancer because telomeres would never shorten to the point at which senescence would occur? Experimental
inducement of cellular immortality (cancer) is difficult to accomplish in human cells by other than specific viral
vectors (Shay, et al, 1991, pp. 2, 6). The in vitro transfection of human cells with T-antigen of the SV40 virus
enables replication to continue past the normal point of senescence until cell crisis occurs, but immortalization of
these cells is extremely rare (Shay, et al, 1991, p. 3). However, rodent cells are more easily immortalized with the
SV40 T-antigen (Shay, et al, 1991, pp. 2-3, 6). Determination of the cause of this difference between human and
rodent cells could elucidate the mechanism by which some cells undergoing cellular crisis are transformed into
immortal cancer cells. The relative ease with which rodent cells are immortalized suggests that experiments in life
extension by maintaining telomere length might better employ a different laboratory animal than rodents.
A potential problem in experimentally lengthening telomeres is that such modification may have an adverse effect on
the organism. Tetrahymena in which telomeres were lengthened appeared sick; however, this effect was probably
due to the fact that the nucleotide sequence was also changed (Kipling, pp. 83, 130; Blackburn, p. 571). Cells of
Schizosaccharomyces pombe thrived for many generations after significant lengthening of their telomeres (Cooper,
et al, pp. 745-746).
The question has been raised as to whether Dolly, the Scottish sheep clone, began life with shorter than normal
telomeres because the source of her DNA was from an adult cell which had already undergone many divisions. If her
telomeres are short, she could be expected to exhibit signs of premature aging at some time and to die at an earlier
age than normal. If this is found to be true, cloning could provide a valuable tool for analysis of senescent gene
expression. However, Fossel has postulated that she probably could not have survived until birth unless her
telomeres had been elongated by telomerase early in her development (personal communication). In humans about
half the length of the telomere is expended during fetal development; development in sheep is presumably similar. If
Fossel's interpretation is correct, experimental lengthening of telomeres in laboratory animals would seem to have a
better chance of success. Analysis of telomeres related to the cloning of Dolly should include ova and udder cells of
the DNA donor sheep, ova from Dolly, and cells from other lambs her age. The ova would provide data on the
maximum length of telomeres in their respective sources.
Protection of chromosome ends and the shortening of telomeres during replication are not the only significant
functions of telomeres. In recent research telomeres or associated proteins were shown to play a role in the
coherence of sister chromatids during mitosis (Kirk, et al, pp. 1478-1481). Telomere-telomere association in the
organization of nuclear chromatin has also been discussed by Kipling (pp. 17, 19-20, 25-26, 99-100).
6. What is the mechanism by which telomerase begins to be expressed in some cells which have reached the crisis
stage of division? What mutation has occurred? What causes the mutation? (See comments above on human versus
rodent cell immortalization.)
7. Is there a correlation between telomere shortening, percentage of mitochondrial DNA guanine converted to
8-hydroxy-guanine, and mitochondrial DNA deletion with aging? If so, what are the causative factors which would
synchronize these processes?
The correlation between telomere shortening in human replicative cells and age has been demonstrated (Allsopp, et
al, 10115, 10116; Chang and Harley, pp. 11191-11192), although with some scatter in the data. A closer correlation
exists between replicative capacity and telomere length (Allsopp, et al, 10115, 10116). A similar correlation
between the 8-hydroxy-guanine content of mitochondrial DNA and mitochondrial DNA deletion in human cardiac
postmitotic muscle cells with age has been identified (Hayakawa, et al, 1992, pp. 979-985; Katsumata, et al, p. 106;
Ozawa, pp. 177-178, 183, 188; Hayakawa, et al, 1996, pp. 369-376). Why do these two apparently unrelated
processes, one operating on replicative cells and the other on nonreplicating cells, both display a correlation with
age? If mitochondrial damage to both types of cells is comparable, then perhaps the elements which could connect
the free radical, mitochondrial, and telomere theories of aging might include the following:
a. Reactive oxygen species (free radicals) are generated in the mitochondria as part of the normal metabolic process.
b. Reactive oxygen species cause somatic mutations and deletions of mitochondrial DNA.
c. Mitochondrial DNA damage causes an increase in the production of reactive oxygen species.
d. Excess reactive oxygen species contribute to cellular apoptosis and necrosis.
e. Cells suffering the greatest damage from free radicals are selected for apoptosis.
f. Apoptosis and necrosis of cells result in replication of other cells to replace them.
g. Replication of cells results in shorter telomeres.
h. Both telomere shortening and mitochondrial damage may participate in the aging process.
That reactive oxygen species damage mitochondrial DNA should come as no surprise considering their potency and
proximity to mitochondrial DNA. Oxidative damage to mitochondrial DNA has been estimated to be at least 10
times as high as to nuclear DNA (Ames, Shigenaga, and Hagen, p. 165; Linnane, et al, p. 643; Ozawa, p. 184). "The
higher levels of oxidative damage and mutation in mtDNA have been ascribed to 3 factors: (a) location of the DNA
near the inner mitochondrial membrane sites where oxidants are formed, (b) lack of protective histones, and (c) lack
of DNA repair activity" (Ames, Shigenaga, and Hagen, pp. 165-166). However, some evidence of a mitochondrial
DNA repair mechanism has been reported (Driggers, et al, pp. 22042-22045; Ames and Shigenaga, p. 88). Ozawa
describes the "vicious cycle" of reactive oxygen species causing mitochondrial DNA damage, which then results in
an increase in production of reactive oxygen species (Ozawa, pp. 185-186).
Apoptosis has been shown to occur as a result of exogenous oxygen stress (Yoneda, et al, pp. 723-729).
Endogenous mitochondrial oxidants are also believed to play a role in apoptosis (Slater, et al, pp. 60-61;
Korsmeyer, et al, 64-65). Ozawa states that ". . . the electron-transport chain could be regarded as the efficient plant
of hydroxyl radical production, essential for the programmed cell death, apoptosis . . ." (Ozawa, p. 182; see also p.
188). Experiments in which human leukemia HL-60 cells were treated in vitro with peroxynitrite stimulated the
formation of reactive oxygen species which then caused apoptosis (Lin, et al, pp. 115-119). Treatment of human
fibroblast cells with H2O2 caused ". . . morphological changes associated with apoptotic cell death . . ." (Yakes and
Van Houten, p.518). The Bcl-2 and Bax proteins are involved in control of apoptosis, with Bcl-2 being commonly
located in the outer mitochondrial membrane (Korsmeyer, et al, pp. 63-66) near the primary source of reactive
oxygen species. The processive pathway by which endogenous free radicals induce apoptosis has not been defined.
One possible course would be to affect the p53 gene which is sensitive to oxidation and which is involved in
apoptosis. However, in an in vitro study done by Russo et al diethylmaleate (DEM) was employed to deplete
intracellular reduced glutathione and hence cause an increase in endogenous free radicals. The effect was to
deactivate p53 and to induce expression of p21 (WAF1/CIP1), which resulted in cell cycle arrest. A number of cell
lines were employed in their experiments, none of which were normal wild type. Use of an unmodified cell line with
an extended range of concentrations of DEM could provide a valuable extension of their approach. Perhaps a lower
dose of DEM would not have deactivated p53, which then might have induced apoptosis. Tumour necrosis factor-a
(TNF-a) is an extracellular inducer of apoptosis (Hannun, p. 1856). Richter et al state that "TNF-a stimulates
mitochondrial ROS [reactive oxygen species] production in mouse fibrosarcoid cells and causes their apoptotic
death. Apoptosis is at least in part related to ROS-induced mitochondrial Ca2+ cycling . . ." (Richter, et al, p. 69).
"These findings pose the question whether one common (final?) pathway of apoptosis is oxidative stress coupled to
mitochondrial Ca2+ cycling" (Richter, et al, p. 72).
Simple logic would suggest that the greater the damage to a cell, the more likely that it will be the victim of
apoptosis. As free radicals damage mitochondrial DNA, the ability of the mitochondria to perform their metabolic
function is reduced, and they then generate even more free radicals. Those cells producing the most free radicals
would be the most likely to be destroyed by apoptosis, whereas those suffering the least damage would be more
likely to survive. This natural selection process would be a significant factor in limiting the accumulation of
mitochondrial DNA damage in replicative cells. Once a cell reaches senescence the selective process ends, and
mitochondrial DNA damage would be expected to accumulate because senescent cells are resistant to apoptosis
(Campisi, 1996, p. 498). Assuming postmitotic cells are also resistant to apoptosis, the process of selecting for less
damaged cells cannot function, and mitochondrial DNA damage accumulates. "Especially in post-mitotic cells, the
mitochondrial genome mutations accumulate during life by escaping the excision repair and cellular selection by
mitosis" (Ozawa, p.178).
Mitogenic factors include oxidants produced by phagocytes and neutrophils in the inflammatory response to
exogenous irritants (Ames and Shigenaga, pp. 88-89). Perhaps mitochondrial oxidants produced during cellular
respiration and which are involved in apoptosis are also mitogenic. If so, is the mitogenic effect of free radicals
direct on the cell in which they are generated, or is it indirect by causing apoptosis of the cell in which they reside?
How do free radicals cause apoptosis? Does the apoptosis then have a mitogenic effect on neighboring cells? What
is the signaling pathway by which other cells are induced to divide? Mordechai et al found that the rate of apoptosis
increased with the age of the individuals tested and that a higher percentage of peripheral blood mononuclear cells
were in the G2 or M phase of the cell cycle in older individuals than in younger (Mordechai, et al, p. 28), indicating
that these cells were in the process of dividing. Division of cells to replace dead ones is a likely occurrence. In recent
research non-mitochondrial reactive oxygen species (primarily superoxide) was shown to be a mediator of
mitogenesis initiated through the Ras gene (Irani, et al, pp. 1649-1652). The study did not address the issue of
whether reactive oxygen species could induce mitogenesis independent of the Ras pathway. Mild oxidative stress
has been reported to increase "rates of telomere shortening and accelerated entry into senescence" (Shay, p. 381).
Shorter telomeres would obviously result from cell division due to the end replication problem. Of course, other
mitogenic factors also affect the number of cellular divisions which occur. The proportion of mitogenic effect which
is caused by free radicals (if any) as compared to other mitogenic factors significantly affects the strength of the
linkage between the mitochondrial and telomere theories of aging postulated here.
For discussions of the telomere theory of aging refer to Shay; Harley, et al; Campisi, 1996; and to Fossel in the
bibliography. The mitochondrial theory of aging is presented by Linnane, et al; Ozawa; Ames and Shigenaga; Ames,
Shigenaga, and Hagen; and Wallace, et al, pp. 141-143, 149.
The course of events listed above might help to explain the parallel results between the ostensibly independent
processes of mitochondrial DNA damage and telomere shortening. The assumption in this hypothesis is that
replicative cells undergo similar mitochondrial DNA damage to that in postmitotic heart muscle cells, but this is not
known. A lower rate of accumulation of mitochondrial DNA damage would be expected in replicative cells due to
the selective removal of damaged cells by apoptosis and the preferential retention of cells in better condition.
However, possibly a lower rate of mitochondrial damage in some replicative cells could be due simply to a lower
metabolic rate than that in heart and brain cells. It would seem unlikely that the two significant aging factors of
mitochondrial damage and telomere shortening could occur simultaneously without interacting in some manner.
Campisi has suggested that ". . . renewable and postmitotic tissues may age by different (albeit interacting)
mechanisms" (Campisi, 1996, p. 499, emphasis added).
However, there may be no direct correlation between mitochondrial aging and telomere aging other than evolution.
There would be no advantage for a species to have telomeres which could last for 500 years if its mitochondria
could only survive for 129 years, or vice versa. The two processes might have no causal relationship other than
evolutionary selection. The 129 year figure is taken from Ozawa (p. 185; see also: Hayakawa, et al, 1992, pp. 980,
982-983; Hayakawa, et al, 1996, pp. 370-371) who states that the ratio of mtDNA with a 7.4 kbp deletion to the
total mtDNA in heart muscle tissue increases exponentially with age up to 7 percent at age 97, the last age for which
data are available. The formula is:
log (deleted mtDNA percentage) = 0.0407 (age) - 3.23
with a correlation coefficient of r = 0.87 at p<0.01. Linear extrapolation of this log function to 100 percent deletion
occurs at age 129. This age would exceed the maximum life expectancy for humans regarding their myocardial
bioenergetic function (Ozawa, p. 185) because an individual would expire before deletion reached 100 percent.
More data are needed for the 95+ year age group to improve the reliability of the extrapolated maximum age.
Additional data also would be expected to exhibit more variation than is shown in the work of Ozawa, Hayakawa,
and their colleagues.
The degree of mitochondrial damage is probably a direct function of the quantity of reactive oxygen species
produced, which in turn can probably be directly correlated with the quantity of energy (ATP) produced and
consumed by the cell as adjusted for the effects of antioxidants. However, as mitochondrial DNA damage occurs the
efficiency of the mitochondria degenerates, and more free radicals are generated in proportion to the ATP produced.
This is what Ozawa calls the "vicious cycle" (Ozawa, pp. 185-186). If there is a correlation between mitochondrial
damage and telomere loss, it would most likely be found in replicative cells which have a high energy consumption.
Consideration should be given to identifying this type of cell and analyzing it for both mitochondrial damage and
telomere shortening. Possible candidate cells could include glial cells in the brain, lung fibroblasts, and intestinal
cells. Because mitochondrial studies have been performed on brain neurons (Ikebe, et al, pp. 1044-1048; Wallace, et
al, p. 149), investigation of associated glial cells could prove informative.
Fossel properly emphasizes the role of telomeres in aging, but states that "The genes in your mitochondria . . . can
be ignored for our purposes" (Fossel, p. 15). He gives due consideration to the effects of free radicals (pp. 26-31,
36-40, 46-47), but suggests that, subject to the inaccuracy of the estimates, "we could expect to die of
mitochondrial loss in about 7,000 years" (p. 215). "Perhaps the damage that accrues depends upon changes in gene
expression in the cell's nucleus and hence upon telomeres that alter that gene expression in aging. In short,
lengthening the telomere might prevent mitochondrial damage" (Fossel, p. 215). Support for this view can be found
in the fact that of the approximately 700 proteins contained in mitochondria, less than 10 percent are transcribed by
mitochondrial genes, the majority being the products of nuclear genes (Kleinsmith and Kish, pp. 512, 658).
Mitochondria are thought to have originated from eubacteria which had developed a symbiotic relationship with
early eukaryotes at least a billion years ago (Kleinsmith and Kish, p. 665). Since then evolution has resulted in
nuclear genes dominating most mitochondrial functions. Whether any of these nuclear genes changes expression
with shortening telomeres would require a more extensive literature search than has been conducted here, and
would more likely require original research. Possibly there is a feedback mechanism of senescent gene expression
affecting mitochondrial respiration in replicative cells, but a more direct connection would seem to be that telomeres
become shorter as a secondary effect of the action of reactive oxygen species primarily in mitochondria. The flow of
cause and effect is most likely from mitochondria to telomeres, rather than the other way around. That changes in
gene expression of senescent replicative cells could produce proteins which would directly cause mitochondrial
DNA damage in other nonreplicative cells is highly improbable. The mitochondrial DNA damage in cardiac muscle
cells which has been correlated with age obviously could not have been caused by telomere induced senescence
within the same cells because the cells are postmitotic and have not divided a sufficient number of times to become senescent.
Fossel has expressed the view that the exponential increase in mitochondrial DNA damage in the later years of
human life is probably triggered by senescent gene expression having a negative effect on mitochondrial function in
proliferative cells (personal communication). He also makes a reasonable case for changes in replicative cells having
a deleterious effect on dependent tissues composed of nonreplicative cells (Fossel, pp. 127-128, 186; see also Chang
and Harley, pp. 11190-11194). Could this relationship account for mitochondrial damage in the latter? For example,
could decreased blood flow to the heart due to atherosclerosis cause greater free radical induced damage to the
heart muscle cells? This example may represent a special case which is a complex interaction beginning with free
radicals causing damage to endothelial cells of the arteries, shortened telomeres due to proliferation of those cells,
inability to repair the damage due to cellular senescence, and atherosclerosis which affects the heart, with possible
increase in reactive oxygen species in the heart. A similar effect may be caused by microvascular disease. However,
in cases of damage to postmitotic cells in tissues which depend on proliferative cells, the initial factor is free radicals,
not senescent gene expression.
In Fossel's opinion significant accumulation of mitochondrial damage would not occur without senescent gene
expression (personal communication). He believes that the cellular germ line has descended from the beginning of
life, or at least since the beginning of mitochondrial symbiosis in eukaryotes, without accumulating mitochondrial
DNA damage. He points to the lack of damage in unicellular life. To a certain degree age is practically meaningless
in one-celled organisms: daughter cells are really the same age as their mother cells. He views multicellular life as a
continuum of unicellular life, so that in a sense we are all about 3.5 billion years old. He asks, ". . . if mitochondrial
damage goes up exponentially, then why does it do so 'instantly' after incurring essentially no damage for the first
3.5 billion years" (personal communication). According to Fossel the mitochondrial DNA damage which
accumulates in the lifetime of individuals now is due to the initiating effect of senescent gene expression which
results from telomere shortening.
A differing view would be that mitochondrial DNA damage has occurred since the first mitochondrion came into
existence. To think that the germ line has continued pristine and inviolate since the beginning of life is a
misconception. DNA damage, mutation, and evolution are all part of the same process. The DNA damage does not
seem to have accumulated due to the Darwinian process of natural selection. Linnane et al pointed out that "The
accumulation of mtDNA mutations in the reproductive germ cell line would also probably result in the formation of
non-functional gametes, or non-viable zygotes after fertilisation" (Linnane, et al, p. 644). Mitochondrial DNA
damage in the germ line is usually not transmitted beyond the first generation because of the poor health of the
individuals inheriting such mutations. Inherited mitochondrial mutations do continue to occur as demonstrated by
the existence of primary mitochondrial diseases (Luft, pp. 1-6; Scholte, pp. 161-191). Mitochondrial DNA
mutations were found to occur in yeast at a much higher rate than in nuclear DNA. The petite mitochondrial
mutation occurred at a rate of 10-1 to 10-3 compared to 10-14 to 10-16 for nuclear DNA (Linnane, et al, p. 643). The
high rate of mitochondrial DNA damage that is exhibited in human postmitotic cardiac cells is not a recent biological
innovation. This type of mutation occurs in more primitive forms of life, and always has. Unicellular life which
inherits mitochondrial mutations does not survive unless the mutation is advantageous or at least permissive. A
significant difference between unicellular and multicellular organisms is that in multicellular life cells with damaged
DNA may still allow the organism to continue to exist due to the support of other less damaged cells. This support
allows some cells to accumulate mitochondrial DNA damage during the life of the individual until the damage is too
great for the organism to survive. On the other hand if a one-celled organism suffers too much damage, it dies. If a
strain or species accumulates too many disadvantageous mutations, the strain or species becomes extinct. Successful
evolutionary mutations have obviously occurred because human mitochondria are not the same as in one-celled
eukaryotic life forms. The fact that most mitochondrial proteins are controlled by nuclear genes is additional
evidence of evolutionary change and that the germ line has not continued without alteration. Perhaps in mammals
inherited mitochondrial DNA damage is minimized because: (1) mitochondria are inherited only from the female
parent; (2) the female's ova are produced while she, herself, is still a fetus; (3) metabolic activity in the ova might be
minimal until fertilization; (4) production of free radicals might also be reduced; and (5) the function of the gamete
or the viability of the zygote would be compromised by damaged mitochondrial DNA. The validity of the third and
fourth items is subject to confirmation. At the same time as ova are presumably dormant, other cells are
metabolically active and are accumulating mitochondrial DNA damage.
Cancer cells have reached and passed the point of cellular senescence. Do they exhibit senescent gene expression? If
so, have they incurred significant mitochondrial DNA damage as would be expected if senescent gene expression is
the primary cause of such damage? If they demonstrate the effects of senescent gene expression but do not contain
significant mitochondrial DNA damage, the postulation that mitochondrial DNA damage in aged individuals is
caused by senescent gene expression would be subject to question. However, when cancer cells avoided cellular
senescence they may have also avoided senescent gene expression. It is also possible that due to the lack of the
selective process of apoptosis mitochondrial DNA damage might accumulate in cancer cells with or without
senescent gene expression.
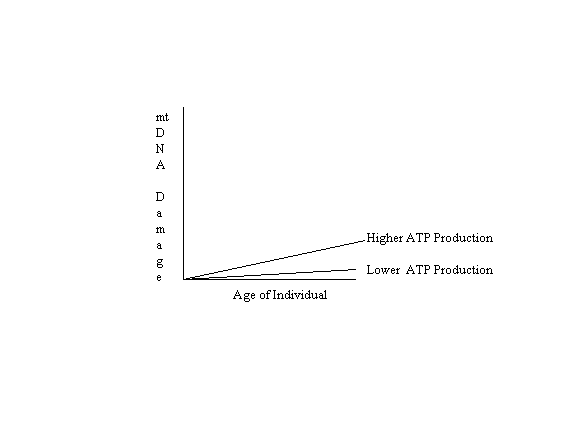
If mitochondrial DNA damage caused by free radicals were accumulated with age solely in proportion to the
quantity of ATP produced, without modification by other factors (antioxidants, the free radical multiplying effect of
DNA damage, mitochondrial dysfunction due to senescent gene expression, and viable cellular selection due to
apoptosis), the following graph would illustrate the
results:
Naturally the other factors play a significant role so that the following graph is more representative of reality except
for the exclusion of apoptotic selection (SGE = Senescent Gene
Expression):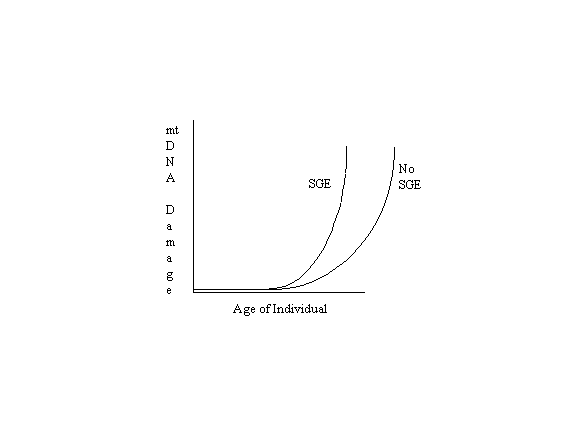
This illustration is similar to published graphs based on actual data (Ozawa, p. 183; Hayakawa, et al, 1992, p. 982;
Hayakawa, et al, 1996, p. 375; Katsumata, et al, p. 106). The two curves represent the accumulation of
mitochondrial DNA damage with age with and without the effects of senescent gene expression (SGE), if any, on
mitochondrial function. If these effects are of major significance, then the curve on the right should be pushed
farther to the right, and it should rise at a slower rate. If, however, the effects of senescent gene expression are of
lesser significance, then the two curves should be closer together or merge as one. Logic alone cannot determine
which of the interpretations presented here is valid. Laboratory research and experiments are needed to answer the
questions raised.
In the literature reviewed those studying mitochondrial damage say little or nothing about telomeres or cell
replication, and those investigating telomeres say little about mitochondrial damage. Research is needed to
determine the relationships between both areas. Most of the research on mitochondria seems to involve the
terminally differentiated cells of heart muscles, brain neurons, and diaphragm muscles. What mitochondrial damage
occurs with increasing age in replicative cells such as fibroblasts? Does it correlate with changes in telomere length?
Both kinds of analyses should be performed on the same cell sample from the same subject. As replicating cells
approach senescence the rate of cell division decreases and finally stops (Cristofalo, et al, p. 429). Could this slow
down in cell division be due to decreased output of energy in the form of ATP from mitochondria which are
becoming dysfunctional? What mitochondrial damage is found in "immortal" HeLa cancer cells donated by Henrietta
Lacks more than 45 years ago? These cells are immortal due to the expression of telomerase, but if mitochondrial
DNA damage is a function of age, how could cancer cells be immortal? Do cancer cells show mitochondrial DNA
deletion? Perhaps some cancer cells seem to be immortal because their energy requirements are much lower than
that of heart muscle cells, and therefore they generate fewer free radicals to cause mitochondrial DNA damage.
Does mitochondrial damage occur primarily in non-replicative cells which have high energy requirements (i.e., heart
and brain cells)?
An in vitro research project which could help answer some of the questions raised here would include the following
objectives: (1) to determine if mitochondrial damage occurs in proliferating cells at the same rate as in quiescent
cells; (2) to determine if mitochondrial damage occurs at the same rate in quiescent cells as in postmitotic cells; (3)
to determine if the production of free radicals is directly proportional to the production of ATP until significant
mitochondrial damage occurs; (4) to determine if the generation of mitochondrial damage is directly proportional to
the production of free radicals; (5) to determine if the available reactive oxygen species increase when the
production of endogenous antioxidants is suppressed; (6) to determine if mitochondrial damage increases when
antioxidants are suppressed; (7) to determine if the rate of apoptosis increases with an increase in mitochondrial free
radicals; (8) to determine if the rate of cell division increases with an increase in mitochondrial free radicals; (9) to
determine the proportion of mitogenic effects due to free radicals; (10) to determine if loss of telomere length is
directly proportional to the amount of mitochondrial free radicals produced; and (11) to determine if the senescent
gene expression resulting from shortened telomeres causes an increase in the production of mitochondrial free
radicals and mitochondrial DNA damage.
The final objective listed would be particularly difficult to accomplish. If senescent gene expression causes an
increase in the production of free radicals, these and mitochondrial DNA damage would appear in senescent cells.
However, mitochondrial DNA damage and the resultant increase in free radical production would be expected to
occur because the damage-limiting process of apoptotic selection would no longer pertain. How can the two
possible causes be distinguished? If senescent gene expression causes an increase in free radical production, then
senescent cells should exhibit a higher rate of generation of free radicals than quiescent cells. Unfortunately,
considering the studies of Ozawa and his colleagues at the University of Nagoya, such observations might require 90
years to obtain results, thereby taxing the patience of any researcher. If the process is accelerated by suppressing the
quantity of endogenous antioxidants, the resulting increase in free radicals might mask any difference due to the
factors of interest. From analysis of mitochondrial DNA damage in apoptotic cells and the rate of apoptosis a
calculated value for damage in senescent cells could be derived, but the problems of time or the masking effect of an
accelerated approach persist. Studies need to be conducted to determine if there is a decrease in the quantity of
mRNA for mitochondrial proteins encoded by nuclear DNA in senescent cells. Proteins which are most likely to
affect metabolism and free radical generation should be given priority. A significant change in such proteins with the
onset of cellular senescence would be prima facie evidence for senescent gene expression being the cause of
putative mitochondrial DNA damage accumulating in senescent cells. Lack of such a change would indicate the opposite.
Parameters for the research project should include: (1) all experiments should use human fetal or neonatal cells
which are cloned from the same source; (2) fibroblast cells would probably be appropriate and would have the
advantage of having been used extensively in other research; (3) human cardiac muscle cells could be used for
experiments requiring terminally differentiated cells; (4) the conditions for each experiment should be identical to all
others with respect to temperature, pH, and nutrients, except as noted; (5) all experiments should include analyses
of telomere length, mitochondrial damage, rate of production of reactive oxygen species, endogenous antioxidants,
rate of production of ATP, rate of apoptosis, and rate of cell division; (6) the analyses listed should be performed at
regular intervals several times during the tests, not only at the beginning and end. Mitochondrial damage in the
control groups is expected to be minimal or even undetectable. Alteration of mitochondrial guanine to
8-hydroxy-guanine in cardiac muscle cells only reached about 1.5% in a 97 year old, and mitochondrial DNA
deletion reached about 7% in the same individual (Ozawa, pp. 183-185). The proposed tests are not expected to last
as long. The experiments to be performed should include the following:
a. Control group 1. These cells should be allowed to proliferate under optimal growth conditions until they reach senescence and finally death. Measurement of the rate of cell division at several stages in this group is important. Production of ATP should be optimal. Any change in production of mitochondrial free radicals after cellular senescence should be identified if possible. Mitochondrial DNA damage should be compared before and after cellular senescence.
b. Control group 2. These cells should be maintained under identical conditions as control group 1, except that all cell division should be prevented.
c. Control group 3. These cells should be maintained under identical conditions as control group 1, except that proliferation should be permissive. All mitogenic factors should be inhibited except for the effect of free radicals. There is a very fine distinction between this group and control group 2. An objective of tests described below will be to determine if free radicals are mitogenic; therefore this control group is needed for comparison.
d. Control group 4. Terminally differentiated cells should be maintained under identical conditions as control groups 1 and 2 with optimal production of ATP.
e. Experiment 1. Cells in this group should be maintained under conditions identical to control group 1, except that production of endogenous antioxidants should be suppressed, with tests performed at several levels. Production of ATP should be identical to that in all of the control groups. The available reactive oxygen species are expected to increase in this experiment as compared to control group 1, which should give accelerated results as compared to in vivo analysis. Mitochondrial DNA damage is also expected to increase. Comparison with control group 1 should also provide data on the relative rates of apoptosis, cell division, and telomere loss. Comparison with experiment 2 should aid in determining if the rate of mitochondrial damage is different between proliferating and quiescent cells.
f. Experiment 2. Cells in this group should be maintained under conditions identical to control group 2, except that production of endogenous antioxidants should be suppressed, with tests performed at several levels as in experiment 1. Production of ATP should be identical to that in all of the control groups. Comparison with experiment 1 should aid in determining if the rate of mitochondrial damage is different between proliferating and quiescent cells. Comparison with experiment 5 should reveal whether the rate of mitochondrial damage in quiescent cells is different from postmitotic cells. Cell division and telomere loss should not occur. The effect on apoptosis, if any, is unknown.
g. Experiment 3. Cells in this group should be maintained under conditions identical to those in experiment 2 except that the rate of production of ATP should be varied in multiple tests. The purpose of this experiment is to determine how the rate of production of reactive oxygen species varies with change in production of ATP.
h. Experiment 4. Cells in this group should be maintained under conditions identical to control group 3, except that production of endogenous antioxidants should be suppressed, with tests performed at several levels as in experiment 1. Note that mitogenic effects other than those caused by free radicals (if any) should be suppressed as much as possible. Assuming that the rate of production of free radicals is directly proportional to the rate of production of ATP (to be determined in experiment 3), the production of ATP should be varied in multiple tests as in experiment 3. Production of free radicals would be expected to increase relative to ATP as mitochondrial damage occurs. The purpose of this experiment is to determine if varying levels of free radicals results in different rates of cell division. If this is found to be true, then extrapolation of the data to a point of production of zero free radicals might help to determine the proportion of mitogenic effects which are due to free radicals as compared to other mitogenic factors in vitro. To determine the relative effect in vivo is more problematic. The effect of free radicals on apoptosis and on telomere loss should also be measured. If possible, these tests should be continued after cells reach senescence to determine the rate of post-senescent mitochondrial DNA damage.
i. Experiment 5. Terminally differentiated cardiac muscle cells should be maintained under conditions identical to control group 4 except that production of endogenous antioxidants should be suppressed, with tests performed at several levels as in experiment 1. Comparison of the results with experiment 2 should help determine whether the rate of mitochondrial damage is different from that in quiescent cells.
If the research project described substantiates the hypothesis outlined earlier, the hypothesis could be described in
the following mathematical terms:
A = quantity of ATP produced/T
C = rate of cell division/T
D = number of mitochondrial DNA deletions/T
E = rate of telomere loss/T
F = free radicals (available reactive oxygen species after effect of antioxidants)/T
G = number of mutations of mitochondrial guanine to 8-hydroxy-guanine/T
I = rate of apoptosis due to factors other than free radicals
L = rate of telomere addition due to telomerase/T
M = non-free radical mitogenic factors as a function of T
N = rate of mitochondrial protein alteration due to senescent gene expression/T
O = quantity of antioxidants in mitochondria during T
P = rate of apoptosis due to free radicals, as number of cells which die/T
R = rate of production of reactive oxygen species/T
S = effect of slowing of rate of cell division as cells approach senescence
T = a convenient but specific unit of time
X = reactive oxygen species neutralized by antioxidants/T
lower case letters = various factors of unknown value which affect the elements indicated
then
R = pA + yG + zD + mN (assuming rate of production of reactive oxygen
species is directly proportional to the rate of production of ATP, plus the added effect of mitochondrial DNA damage, plus the effect of senescent gene expression, if any)
X = qO (neutralized reactive oxygen species is a function of mitochondrial
antioxidants)
F = R - X (net free radicals after effect of antioxidants)
= pA + yG + zD + mN - qO
G = rF (assuming guanine mutations are a function of free radicals)
D = sF (assuming mitochondrial DNA deletions are a function of free radicals)
P = tF (assuming the rate of apoptosis is in part a function of free radicals)
C = uP + vF + kI + wM (assuming the rate of cell division is a function of the
rate of apoptosis plus any direct effect of free radicals plus the effect of other mitogenic factors)
= tuF + vF + kI + wM
= (tu + v)F + kI + wM
E = xC - S - L (telomere loss is a function of the rate of cell division as adjusted
= x((tu+v)F + kI + wM) - S - L for slowing on approach of senescence and
as adjusted for the effect of telomerase, if any)
N = nE (valid if shortened telomeres have a positional effect on expression of
nuclear genes controlling production of mitochondrial proteins)
The next to last formula shows telomere loss to be a function, in part, of the net production of free radicals,
according to the hypothesis described above. A diagram showing the key elements of the hypothesis follows:
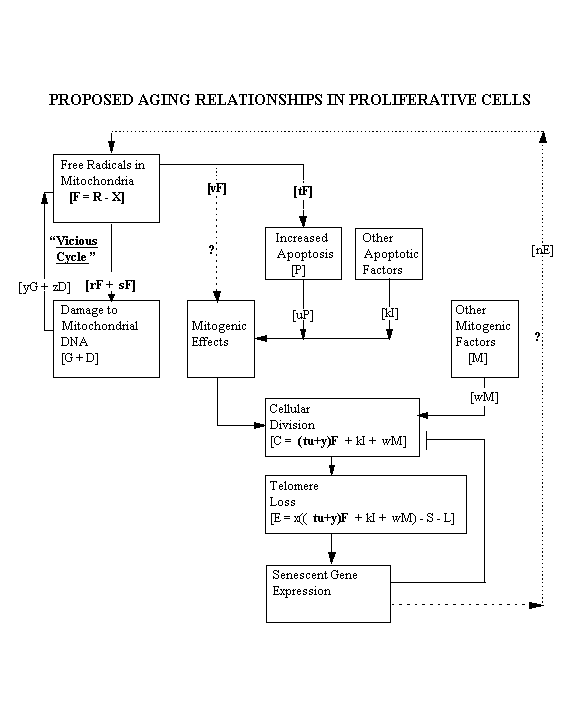
[Elements of this item are similar to elements of Dr. L. Stephen Coles's item number 5.]
8. What mechanism(s) exist for the repair of mitochondrial DNA damage? Can therapeutic intervention promote this repair?
Evidence of a mitochondrial DNA repair mechanism has been reported (Driggers, et al, pp. 22042-22045; Ames and
Shigenaga, p. 88), but elsewhere Ames, Shigenaga, and Hagen refer to a lack of a mitochondrial DNA repair
mechanism (pp. 165-166; see also Linnane, et al, p. 642). Yakes and Van Houten found that after exposure of
human fibroblast cells to H2O2 for 15 minutes, mitochondrial DNA was damaged, but was then repaired after 1.5
hours; however, if the exposure was for 60 minutes, the damage could not be repaired. Nuclear DNA experienced
less damage and was more readily repaired (Yakes and Van Houten, pp. 514-519). In novel in vitro experiments
peptide nucleic acids (PNAs) were synthesized to be complementary to human mitochondrial DNA templates which
contained a deletion breakpoint or a single base mutation. The PNA bound to the defective mitochondrial DNA and
prevented it from replicating while normal DNA was able to replicate. This approach provides for the potential
treatment of mitochondrial DNA defects when intracellular heteroplasmy exists (Taylor, et al, pp. 212-215).
9. Would it be feasible to extract mitochondria from cells in which there is little mitochondrial damage, multiply those mitochondria ex vivo, and implant them into high energy cells which have significant mitochondrial damage? Could damaged mitochondria be deleted from the target cells? How could such a procedure be implemented?
Procedures for extracting mitochondria have been described (Kleinsmith and Kish, pp. 137-139, 316). Procedures
for transplanting mitochondria have been described in (Coles, pp. 24-25). When yeast cells are cultured under
anaerobic conditions their mitochondria seem to disappear; when subsequently exposed to oxygen the mitochondria
reappear as promitochondria which can then develop into normal mitochondria (Kleinsmith and Kish, p. 660). If the
same results could be obtained with human mitochondria, perhaps the smaller promitochondria could be more
readily implanted where needed.
10. Is there a relationship between telomere length and mitochondrial DNA damage in progeric patients and
individuals with mitochondrial diseases?
This issue was stimulated by consideration of short telomeres in progerics (Fossel, pp. 116-117) and a study by Takayuki Ozawa and colleagues of a prematurely aged 19 year old male (Katsumata, et al, pp. 102-110). The telomere information comes from Allsopp et al (pp. 10116-10117) and Goldstein (1978, pp. 177-181, 196-200), who was also a co-author in the Allsopp paper. Goldstein's work includes a nine year old progeric which he had previously described (Goldstein, 1969, p. 424) and two others (Goldstein, 1978, p. 198, as cell population doublings). The Allsopp study was limited to five progerics, only one of whom was fetal or newborn (the same individual was apparently also included in the Goldstein study). Data are also available on a girl who was almost 5 years old (Martin, et al, pp. 88-90) and from 11 progerics ranging in age from 1 to 20 years (Brown, et al, pp. 529-532). In general the 19 individuals included in these studies had shorter telomeres than non-progerics, but there is considerable scatter in the data, and it overlaps with that of normal individuals, particularly in the Brown study (Brown, et al, pp. 529-530). The paucity of data for very young children is very evident. Only two were one year old or less, and one of them had nearly normal length telomeres (Brown, et al, p. 530). Allsopp et al list three possible causes: (1) inheritance of shorter telomeres (favored by Fossel, p. 116), (2) "a high rate of cell turnover during development," or (3) "an aberrantly high rate of telomere loss with each cell division" (Allsopp, et al, p. 10116). Allsopp et al eliminated the third possibility in their study, but did not determine which of the other two was preferred. An increased metabolic rate (Goldstein, 1978, p. 179) suggests the possibility of a concurrent increased rate of cell proliferation. At present the cause of shorter telomeres in progerics must be considered as undetermined.
A prematurely aged ninteen year old male was described as having "the remarkable symptoms of progeria, such as
arteriosclerosis, a senile masque, and multiple infarctions in the basal ganglia, which were quite different from those
in patients with mitochondrial encephalomyopathy" (Katsumata, et al, p. 106). They do not specifically state that the
patient was a progeric, but merely point out the similarities to progeria. A symptom which was not in accord with
progeria was deafness (Goldstein, 1978, pp. 177-180; Katsumata, et al, p. 103; Mills and Weiss, p. 89; Brown, et
al, pp. 522-524). The occurrence of convulsions in the patient might also differentiate him from progerics. Goldstein
does describe one progeric who died of convulsions (Goldstein, p. 180), but considering the similarity of symptoms
with some mitochondrial diseases, that person may have been misdiagnosed. Several symptoms of the Katsumata
patient were similar to those of Myoclonus Epilepsy and Ragged Red Fibers syndrome (MERRF), including
deafness, convulsions, and ragged red fibers (Katsumata, et al, pp. 103-104; Luft, p. 2). There is considerable
overlap between mitochondrial syndromes (Luft, p. 2), and Scholte classified more than 120 varieties (Scholte, pp.
161-191). The diagnosis of the 19 year old male and of a briefly mentioned 17 year old female was mitochondrial
cardiomyopathy. Telomere length was not examined in the study, which was limited to terminally differentiated
heart muscle cells.
Both progerics and prematurely aged individuals with mitochondrial diseases need to be studied in a comprehensive
manner that includes telomere length, mitochondrial damage, rate of reactive oxygen species production, antioxidant
profile, rate of cellular apoptosis, and percent of cells in the G2 or M phase of the cycle.
11. Could the following procedure provide a practical method to prevent organ rejection by a potential recipient?
a. Expose a sample of the individual's lymphocytes ex vivo to either a low or high concentration of the antigen(s) of the organ to be transplanted.
b. Reintroduce the multiplied lymphocytes back into the organ recipient.
Exposure of an individual to moderate concentrations of an antigen normally induces an antibody response.
However, if the concentrations are either very low or very high, action of T suppressor cells prevents the formation
of antibodies (Kleinsmith and Kish, p. 741). Perhaps using a procedure analogous to adoptive immunotherapy (see
Old, p. 142), but for the purpose of amplifying T suppressor cells rather than antibody producing lymphocytes,
would be an effective method to reduce immune response without using drugs which would weaken the entire
immune system. If this method were successful, it could also be used to prevent rejection of viral vectors used in a
variety of treatments.
12. Does ingestion of Coenzyme Q-10 extend human life? If so, is there a correlation between dosage and life extension?
It has already been shown that CoQ-10 significantly extendes lifespan in rodents (Coles and Harris, pp. 205-216).
For human trials, large numbers of Japanese have been using Coenzyme Q-10 since the mid 1970's (Bliznakov and
Hunt, p. 2; Lee, p. 25). A demographic study of users versus nonusers of coenzyme Q-10 with appropriate
consideration of dosage, diet, and the exclusion of smokers could prove valuable. Because Coenzyme Q-10 is
known to be beneficial to individuals with cardiac problems, it would be of considerable interest to determine the
relative difference in cardiac mitochondrial damage between deceased persons who had regularly used Coenzyme
Q-10 and those who had not. Telomere length in replicative cells should also be determined.
For convenience secondary sources were employed in the preparation of this list. Primary sources are obviously preferable.
All of the topics discussed here need a much more thorough review of the research literature than was performed
during the writing of this paper.
Researchers should be encouraged to look beyond the narrow focus of their particular interest to include related
factors which in combination could be much more significant than any one factor alone. Such factors as telomere
length, percent of senescent cells, percent 8-hydroxy-guanine content of mitochondrial DNA, percent mitochondrial
DNA deletion, percent of cells in G2 or M phase of the cell cycle, rate of apoptosis, antioxidant levels, and oxidative
stress should be considered in conjunction with the primary subject of the research. Inclusion of these and other
appropriate factors in scientific studies would facilitate correlation of the multiple factors involved in aging.
I would like to thank L. Stephen Coles for his contributions to several sections of this document, for discussing the
concepts presented, for his forbearance in tolerating continuous revisions, and for initially suggesting the project.
Michael Fossel provided stimulating E-mail discussions and suggestions which were very beneficial. Charles
Thomas, Jr. instigated the interest in the mitochondrial theory of aging and assisted with references to the work of Ozawa.